Immune Sensitivity
Q: Why Study the Dynamics of the Immune Synapse?
We want our immune systems to be highly sensitive when a new pathogen arrives or conditions in our bodies change. The Immune Synapse is the name given to the junction between a T cell and an adjacent antigen-presenting cell. In order for T cells to recognize foreign material and initiate an immune response, they use proteins called T cell receptors (TCR) to ‘touch’ pieces of that material (peptides) that are displayed on proteins (MHC) on the surfaces of antigen-presenting cells. We’ve been studying, since the lab’s inception, how T cells physically interact with and detect opposing surfaces. This is the critical moment in immune recognition—the point at which a cell detects the foreign material and can begin the process of cell division and gene-expression that allows it to respond.
Current Projects: Microvilli!
We are currently studying how T cells use small finger-like projects called microvilli as a critical part of their detection. While microvilli were observed via electron microscopy for many years, our ability to watch these tiny structures in real time and see how they work was limited because they were so small and fast-moving. T cells effectively are waving these fingers around over their surface. We solved this deficiency using lattice-light sheet imaging, a technology that we found in Eric Betzig's lab (Thanks Eric!), worked to develop and then import and adapt at UCSF. T cells, scanning their environments look like this (T cell in green):
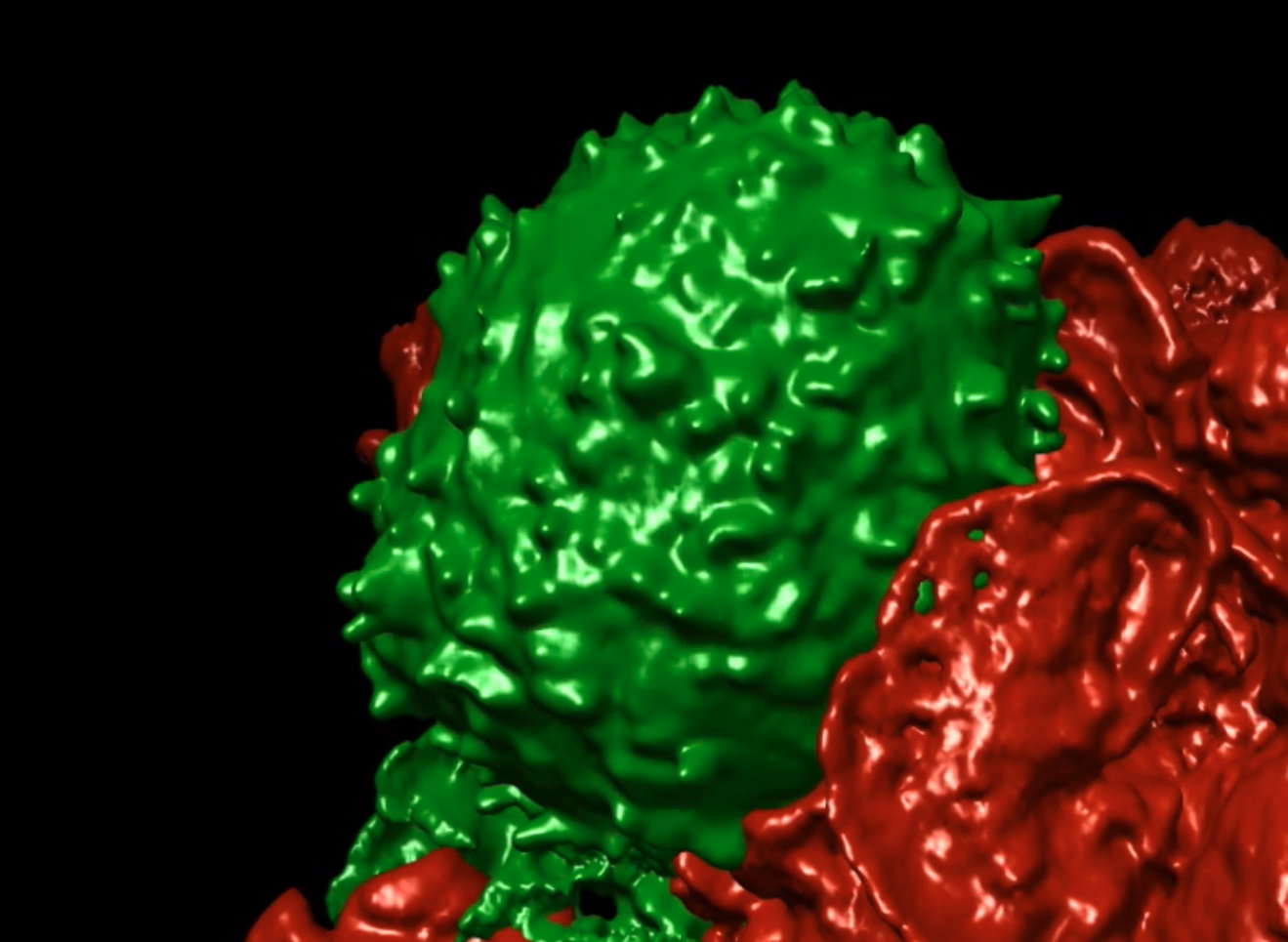
In Cai et al Science 2017, we demonstrated that T cells actively extend, retract and move their microvilli in order to efficiently scan opposing surfaces. The movement allows them to scan an entire surface in about 1 minute despite the observed fact that at any one time, they may only contact a small fraction of the surface on the other side--effectively they are palpating the world outside. We also found that they only touch a given site on the opposing cell for about 5 seconds. That 5-second contact makes us rethink T cell antigen-detection since that timing means that receptors must coalesce on peptide-MHC complexes in that very short period. We also found that TCR engagement solidifies the microvillar projection, seemingly ‘gluing’ the contact in place. That this did not require ZAP-70 signaling or the actin cytoskeleton implies that the TCR ‘locks-in’ synaptic contacts without the need for cellular signaling. We are now looking to understand how membrane proteins aggregate into the various 3D regions of the T cell and how that allows T cells to be both sensitive and also not over-reactive.
Historical: Tracking T cell receptors and co-receptors during engagement.
As a postdoc and just prior to starting our lab, I made the first fusions between GFP and TCR components and expressed them in live T cells. By simultaneously visualizing calcium levels in T cells, we were able to show that signaling onset occurred prior to cSMAC formation and in a phase characterized by submicron cluster formation. We also showed differential movement patterns of CD3 and CD4 components, suggesting that these two proteins come together and fall apart during the process of recognition.(Krummel et al. Science 2000)
In Moss et al. PNAS 2002, we adapted segmentation algorithms and 3D quantization approaches to determine the velocities of membranes and receptors during the first moments of T-APC contact. This led us to be able to conclude that receptor movement was active, approximately 0.1-0.2um/min and not likely achieved via diffusion alone. It also revealed a membrane wave that initiates during the T-APC contact. The methods for mining data out of 3D datasets have been repeated in other systems by other groups and applied to similar approaches of membrane deformation.
We subsequently assessed costimulatory ligands CD28 (Andres et al. 2004) and CD40L/CD40 pair (Boisvert et al 2004) and found that both are recruited to the immunological synapse coincident with when clusters of TCR were forming but with different kinetics: CD28 appears in the earliest phases of our observations whereas CD40L/CD40 arrive after and likely as a consequence of the polarization induced by TCR signaling.
To define TCR dynamics directly, as opposed to simply the associated CD3 chains, and as a means to analyze receptor dynamics in unmanipulated naïve T cells, we generated TCR transgenic mice expressing only T cells with a variant of the ovalbumin (OVA) reactive in which the TCR alpha chain is fused to eGFP (Friedman et al. JEM 2010). Direct imaging of TCRs during their interactions with dendritic cells in this context revealed that the most reliable common theme for responses to T cell stimuli was the observation of rapid TCR-GFP internalization—unlike with CD3-GFP, the TCR-GFP appears to persist inside these cells, allowing us to continue to track the receptors. TCRs when observed in these mice, appear to become flexibly arrayed into the synapse, often without forming a centralized ‘cSMAC’ at all but through a process of apparent sequential receptor triggering and internalization. Synapse formed by these cells also often continue to actively move whilst signaling continued motility, largely divorcing motility arrest as a pre-requisite for signaling; a feature of activation of T cells in situ highly suggestedfor cells in lymph nodes by Cahalan and van Andrian’s labs. Using the TCR-GFP T cells directly in lymph nodes, we were then able to study TCR dynamics in vivo through optimization of our 2-photon detection and signal processing. Using the readout of internalized TCR-GFP vesicles as a readout of T cell activation, we once again observed rapid internalization in the absence of either motility arrest or evident cSMAC formation.
This work was extended in Beemiller et al Nature Immunology 2012 in which we demonstrated the concurrence of signaling T cell receptor microclusters on the T cell surface and how this type of signaling can concur in time with ongoing motility as a transient synapse is formed. We also demonstrated how actin movements are utlized to coordinate these two seemingly-disparate activities. Using a life-act probe, we also demonstrated that formation of a cSMAC requires an active actin depolymerization at the center of the synapse.
The sum of these studies to date suggests that assembly of TCR clusters does not rely on a ‘stable’ cSMAC and can take place against a background of motility. The details of how signaling clusters initially organize or are perpetuated in this setting is yet to be understood.
Other Cytoskeletal Players:
We have also used image-based screening to isolate gene-products important for T cell function as well as for motility (see Tooley et al. Seminars in Immunology. 2005. In Tooley, Gilden et al. Nature Cell Biology. 2008, our lab was the first to identify the Septin cytoskeleton as an important player in T cell cortical tension and for effective motility. While septin fibers appear to form a corset around the midbody of T cells and thus reinforce this zone, the septin cytoskeleton also appears to reinforce cortical rigidity and tension. This results in two distinctive phenotypes: First, a ‘floppiness’ that results in an elongated uropod that does not track the cell body and the second is an ability to generate or maintain excess protrusions. We hypothesize that this latter feature accounts for our demonstration that septin-null T cells had gained the ability to transit very constrictive barriers, a feature perhaps quite important both for immune tissue surveillance and in cancer metastasis. When we then made septin knockouts, we found that these proteins are also specifically required for cell division when T cells are no anchored to other cells. Cell division while in contact with an APC was septin-independent. We hypothesize that specific septin inhibitors would block cell division to soluble cytokines in vivo while sparing responses mediated by synaptic engagements.